Discovery
Medicinal Chemistry Services
Discovery ADME
- Metabolic Stability
- CYP Interaction
- Permeability & Transporters
- Physicochemical Properties
- Protein Binding
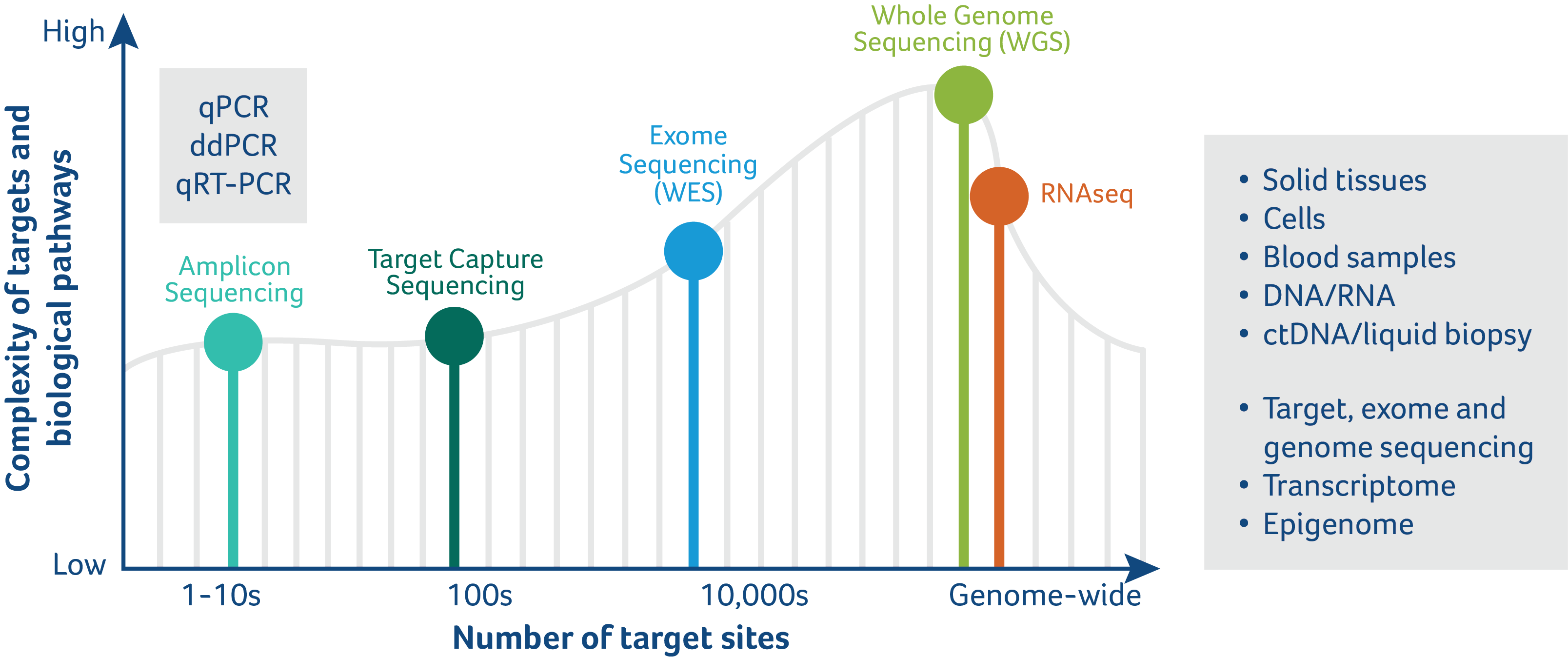
Overview of Frontage’s Complete Genomics Services
Frontage offers cost-effective examination of oncogenes from tumor tissue, biofluids, or genomic or cfDNA samples using targeted sequencing of oncopanels developed by industry leading manufacturers.
Learn MoreOur RNA sequencing services use a combination of state-of-the-art equipment that provides quick and accurate results for your RNA profiling projects.
Learn MoreFrontage offers advanced sequencing technologies that enable profiling of the microbiome DNA composition and the microbiome RNA composition at unprecedented scales
Learn More
Frontage Laboratories websites use cookies. By continuing to browse the site you are agreeing to our use of cookies. For more details about cookies, and their use, please see our privacy policy.
Accept Privacy Policy